 Home
Home
| VOLUME 88 (2008) | ISSUE 11 | PAGE 844 |
|---|
|
Density-functional calculation of the Coulomb repulsion and correlation strength in superconducting LaFeAsO
V. I. Anisimov, Dm. M. Korotin, S. V. Streltsov, A. V. Kozhevnikov+, J. Kunes*, A. O. Shorikov, M. A. Korotin
Institute of Metal Physics RAS, 620041 Yekaterinburg GSP-170, Russia +Joint Institute for Computational Sciences, Oak Ridge National Laboratory Oak, TN 37831-6173 Ridge, USA *Theoretical Physics III, Center for Electronic Correlations and Magnetism, Institute of Physics, University of Augsburg, 86135 Augsburg, Germany ID: 71.45.Gm, 74.25.Jb
Abstract
Constrained density functional theory scheme in Wannier functions formalism has been used to calculate Coulomb repulsion U and Hund's exchange J parameters for Fe-3d electrons in LaFeAsO. Results strongly depend on the basis set. When O-2p, As-4p, and Fe-3d orbitals are included, computation results in U=3 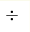 4 eV. With the
basis set restricted to Fe-3d orbitals only, computation gives parameters
corresponding to F0=0.8 eV, J=0.5 eV. Local Density Approximation
combined with Dynamical Mean-Field Theory calculation with these parameters
results in weakly correlated electronic structure. 4 eV. With the
basis set restricted to Fe-3d orbitals only, computation gives parameters
corresponding to F0=0.8 eV, J=0.5 eV. Local Density Approximation
combined with Dynamical Mean-Field Theory calculation with these parameters
results in weakly correlated electronic structure.
|