 Home
Home
| VOLUME 115 (2022) | ISSUE 2 | PAGE 108 |
|---|
|
On the ab initio calculations within DFT + U approach of physical properties of a compound with strong electron-electron correlations by the case of KFeS2
A. G. Kiiamov, M. D. Kuznetsov, R. G. Batulin, D. A. Tayurskii
Department of General Physics, Institute of Physics, Kazan Federal University, 420008 Kazan, Russia
Abstract
The magnetic and electronic properties of quasi-one-dimensional iron chalcogenide compound KFeS2 have been calculated within two DFT + U approaches based on simplified LSDA + U approach and rotationally invariant LSDA + U approach. It is shown that simplified LSDA + U approach with the single 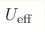 parameter
correctly describes the magnetic ground state but mistakenly predicts the electronic properties. On the other hand,
the rotationally invariant LSDA + U approach with two independent parameters U and J provides the correct description of
electronic and magnetic properties of KFeS2 at once. By the comparison of ab initio results with
experimental data on magnetic ground state and electronic properties the values of U and J parameters have been proposed for the correct description of physical properties of KFeS2 within DFT + U approach. parameter
correctly describes the magnetic ground state but mistakenly predicts the electronic properties. On the other hand,
the rotationally invariant LSDA + U approach with two independent parameters U and J provides the correct description of
electronic and magnetic properties of KFeS2 at once. By the comparison of ab initio results with
experimental data on magnetic ground state and electronic properties the values of U and J parameters have been proposed for the correct description of physical properties of KFeS2 within DFT + U approach.
|